Översikt Cystisk fibros
Cystisk fibros (CF) är den vanligaste svåra ärftliga sjukdomen i den vita (kaukasiska) befolkningen. Det är en multiorgansjukdom med dominerade symtom från de övre och nedre luftvägarna, där infektioner och segt slem utgör de största problemen. Det är en mycket heterogen sjukdom där både symtom, vilka organ som påverkas och när under livet som besvären uppkommer, varierar. Det är en progressiv sjukdom, men tack vare allt bättre behandling har Cystisk fibros (CF) skiftat från att vara en barnsjukdom till att bli en vuxensjukdom med en förväntad medellivslängd på minst 50 år för barn med CF som föds idag (1).
Historisk bakgrund Cystisk fibros
Det finns äldre historiska beskrivningar som skulle kunna vara förenliga med cystisk fibros. Redan 1595 beskrev professor Pieter Pauw i Holland en obduktion av en 11-årig ”förhäxad” flicka som avlidit där man fann en svullen pankreas och cirros i levern. Drygt 10 år senare kom en publikation av Juan Diego, professor i Medicin i Henares i Spanien, där han även använde begreppet förhäxade barn som dog unga och som smakade salt då man gned fingrarna mot deras pannor. Tyska barnvisor från 1700-talet anger att ”det är synd om barn som smakar salt ty de ska dö unga” och man beskrev saltränder på barnens skor. I början av förra seklet började man förstå att det fanns ett samband mellan symtom från mag-tarmkanalen och de från luftvägarna. Den schweiziske barnläkaren Guoido Fanconi publicerade 1936 de allra första kliniska fallen av cystisk fibros hos två barn, 10 mån respektive 3 år, som båda avled. Han påtalade sambandet mellan celiaki, cystiska förändringar i pankreas och bronkiektasier i lungorna. Det publicerades ganska många fallbeskrivningar med barn som hade symtom som sannolikt hade cystisk fibros men ingen hade samlat dessa under någon gemensam beskrivning eller under något gemensamt namn.
Den dansk-amerikanska barnpatologen Dorothy H Andersen var den som gav sjukdomen dess namn. Hon använde ordet cystisk fibros för första gången 1938 i sin artikel ”Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study”. Hon beskrev 49 barn med symtom från luftvägar och magtarmkanal, som avlidit unga, och där hon fann cystiska förändringar i pankreas. Cystisk fibros har således inget att göra med några patologiska förändringar i lungorna.
På 1940-talet ändrade man benämningen till mucoviscidos (segt slem) som är en bättre beskrivning av sjukdomen, men man återgick senare till cystisk fibros. Patologen Sidney Farber från Boston förstod att det rörde sig om en systemsjukdom, dvs påverkan även på andra organ utöver lungor och bukspottkörtel. Han noterade även förändringar på slemhinnan och den negativa inverkan av bakterier i luftvägarna. Redan då tolkade man att det var en recessivt ärftlig sjukdom.
I samband med en värmebölja i New York 1948 drabbades vissa barn av uttalad uttorkning och flera av dem visade sig ha CF. Svettest lanserades 1959, där en förhöjd kloridhalt i svett används för att diagnostisera CF.
Under 1950- och 60-talen kunde man framställa pankreaspulver utvunnet från svinpankreas och tillförsel av detta vid måltid blev närmast en revolution då patienterna kunde äta mat med normal fetthalt och inte enbart fettsnål kost med hög kolhydratinnehåll, och patienterna kunde behålla och öka sin vikt. Ungefär samtidigt utvecklades diagnostik av bakterier och möjligheter att behandla infektioner med förbättrade antibiotika. Dessutom introducerades egenträning, inhalationer och andningsgymnastik för slemmobilisering vilket man i Norden var föregångare med. Tidigare var det vanligt med passiv mobilisering med dränage i olika lägen, vilket numera är helt borta. Under de sista årtiondena av 1900-talet introducerades intensiv antibiotikabehandling för att förhindra kolonisation av bakterier i lungorna och i kombination med kraftigt förbättrad omvårdnad noterade man ökad överlevnad vilket fortsätter för varje decennium.
Patofysiologi
Den kliniska bilden vid Cystisk fibros har varit känd i många år men först 1989 kunde man påvisa den genetiska defekten, som sitter på kromosom 7 (7q31.2) som kodar för jonkanalen CFTR (Cystic Fibrosis Transmembrane conductance Regulator). Den är en port i cellmembran som släpper igenom joner och är uppbyggd av proteinkomplex. CFTR kan öppnas eller stängas under påverkan av elektrisk spänning, läkemedel (se nedan under CFTR-modulatorer), signalsubstanser eller av mekanisk belastning. Vid Cystisk fibros (CF) påverkas transporten av natrium, klorid och bikarbonat, vilket påverkar salt- och vattentransporten över cellmembranet. Den minskade klorid- och bikarbonatutsöndringen och det ökade återupptaget av natrium och vatten i cellerna vid cystisk fibros leder till att ett förändrat, segt och intorkat sekret bildas i luftvägarna.
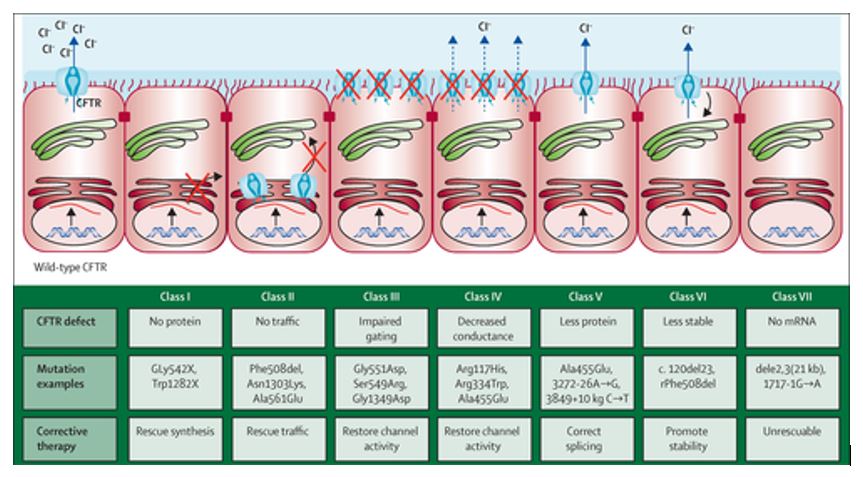
Hittills har man funnit över 2000 varianter (mutationer) på kromosomen och av dessa är cirka 350 säkert associerade med CF medan de övriga är beskrivna vid Cystisk fibros (CF) men är mycket ovanliga. I Sverige dominerar delF508 där cirka 50 % av alla patienter är homozygota, dvs de har två identiska mutationer, dvs en från vardera föräldern. Dessutom har 35 % delF508 och ytterligare en annan mutation, dvs de är heterozygota för delF508 (2). Patienter från länder utanför Europa har helt andra mutationer. Typ av mutation har betydelse för vilket fel som uppkommer då proteinet CFTR bildas. Man brukar dela upp mutationerna i sju klasser varav klass I-III är mer allvarliga och vanligen associerade med pankreasinsufficiens. Klass IV-VI har oftast lindrigare symtom och dessa mutationer är vanligen pankreassufficienta. De utgör cirka 15 % och diagnostiseras vanligen vid senare ålder pga avsaknad av påverkan på pankreas och därmed färre problem med magen och bättre viktutveckling. Prognosen är bättre för dessa med lindriga mutationer. Klass VII har en särställning eftersom den kliniskt är en svår mutation på samma sätt som klass I då inget CFTR bildas. Samma mutation kan ge väldigt varierande symtom. Det finns rimligen många andra modifierande gener/faktorer som styr symtom och sjukdomsutveckling men som vi idag inte känner till.
- Klass I: Inget CFTR bildas
- Klass II: CFTR bildas inte färdigt och endast en liten del når cellmembranet (defekt konfiguration)
- Klass III: Proteinet når cellmembranet men reagerar inte som det ska på öppningssignaler (defekt funktion)
- Klass IV: CFTR-kanalen får nedsatt genomsläpplighet (nedsatt funktion)
- Klass V: Medför för liten mängd normalt CFTR vid cellytan
- Klass VI: Nedbrytningen av normalt CFTR sker för snabbt vid cellytan.
- Klass VII: Inget m-RNA-bildas och därmed inget CFTR.
Figur 1:
Källa: Acta Paediatrica, Volume: 109, Issue: 5, Pages: 893-899, First published: 03 January 2020, DOI: (10.1111/apa.15155)
Förekomst Cystisk fibros
Sjukdomen varierar mycket i världen. I Sverige föds cirka 15-20 barn per år, en incidens på cirka 1/5600 och utan någon könsskillnad. Dock diagnostiseras inte alla barn vid födelsen och det finns sannolikt odiagnostiserade patienter med mycket lindriga symtom som aldrig får diagnosen under sin livstid. I Sverige har vi cirka 750 kända fall. I USA finns cirka 30 000 fall. De länder som har högst förekomst är Irland och UK (1/2500). Som jämförelse är förekomsten mycket lägre i asiatiska befolkningar (1/90 000) och i svarta befolkningar (1/17 000). Lägst förekomst i Norden har Finland med 1/25 000 medan förekomsten i Danmark och Norge motsvarar den i Sverige (3,4,5).
Ärftlighet Cystisk fibros
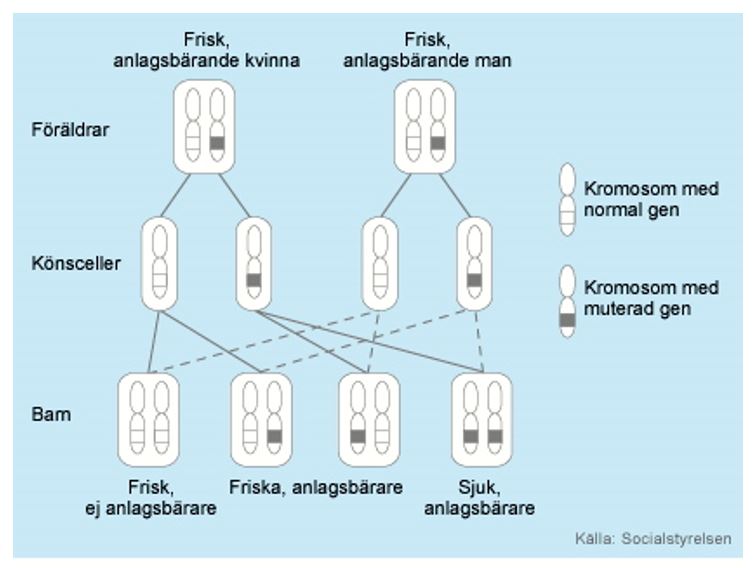
Cystisk fibros är en autosomalt recessiv sjukdom dvs man måste få anlaget från båda föräldrarna, som är friska bärare vilket inte medför några symtom. Vid varje graviditet är det 25 % risk att barnet får Cystisk fibros (CF), 25 % sannolikhet att bli helt frisk och 50 % sannolikhet att barnet blir bärare. En patient med Cystisk fibros (CF) som får barn med en frisk person utan bärarskap överför bärarskapet till samtliga barn. Anlaget för Cystisk fibros (CF) finns hos c:a 2-3 % av befolkningen (6). I familjer är det vanligt att flera syskon har sjukdomen men det är inte enbart de defekta mutationerna som avgör symtomen, som kan variera väldigt mycket mellan syskon, dvs det är en mycket heterogen sjukdom.
Figur 2:
Källa Socialstyrelsen Sällsynta hälsotillstånd 2022
Diagnos Cystisk fibros
En del barn diagnostiseras i anslutning till förlossningen på grund av mekoniumileus, dvs tarmvred på grund av förstoppning, som kan kräva akutoperation (10-15 %) (2). Dessa fall missas sällan men för övriga är det vanligt att det tar viss tid att ställa diagnos, som baseras på diarréer, dåligt viktuppgång och luftvägsbesvär. I Sverige diagnostiseras de flesta barn före ett års ålder. Det förekommer att det tar flera år för diagnosen där man misstolkat symtomen som svårbehandlad astma eller oklara bukbesvär. Det är relativt vanligt att när man diagnostiserar ett nyfött barn med Cystisk fibros (CF) så finner man att det några år äldre syskonet som haft oklara luftvägsbesvär också har Cystisk fibros (CF). I tonåren kan återkommande eller kroniska luftvägsinfektioner (speciellt med ovanliga bakterier), kronisk hosta eller recidivande sinuiter, otiter och näspolyper leda till diagnos. För dem som har få symtom från luftvägarna kan en infertilitetsutredning i 25-30-årsåldern leda till att man diagnostiserar en lindrig Cystisk fibros (CF). De äldsta patienter som diagnostiserats har varit i 70-års åldern och det har skett då man har gjort mutationsanalys pga att yngre släktingar diagnostiseras med Cystisk fibros (CF). Vid noggrann anamnes-genomgång finner man ofta att dessa patienter har haft återkommande om än lindriga symtom från övre och nedre luftvägar. Det finns patienter med typiska symtom inklusive patologisk svettest där man inte kan påvisa mer än en mutation och den andra förblir odiagnostiserad. Dessa får kallas för CF-lik sjukdom eller enbart bronkiektasier om dessa finns men för övrigt bör de behandlas som ”vanlig” Cystisk fibros (CF).
Symtom Cystisk fibros
Symtom (dvs fenotyp) är bara delvis korrelerade till mutation (genotyp) och det gäller speciellt lungfunktion. Det finns stor variation i svårighetsgrad och symtom. Samma mutation kan ge mycket varierande symtom och engagera varierande antal organ även bland syskon. De allvarligaste symtomen kommer från luftvägarna. Eftersom CFTR uttrycks i många organ medför CF i varierande grad symtom från dessa.
Bihålor: Många patienter har återkommande polyper och sinuiter och förtätade bihålor som kräver återkommande operationer och utrymningar. Nästäppa och rinnande näsa är vanligt. Många patienter besväras av återkommande svår huvudvärk. Nära samarbete med öronläkare är viktigt.
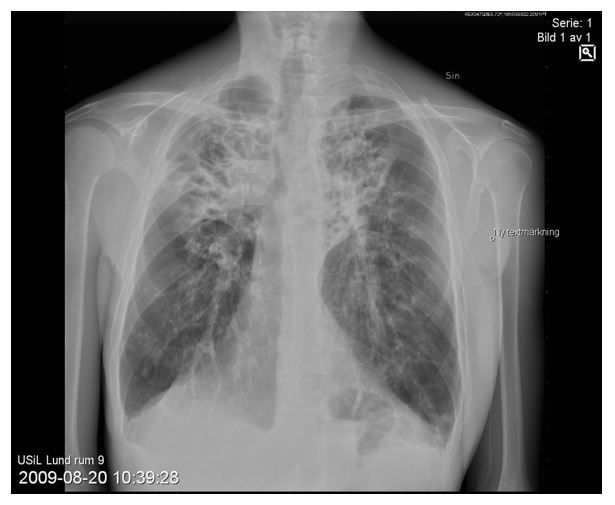
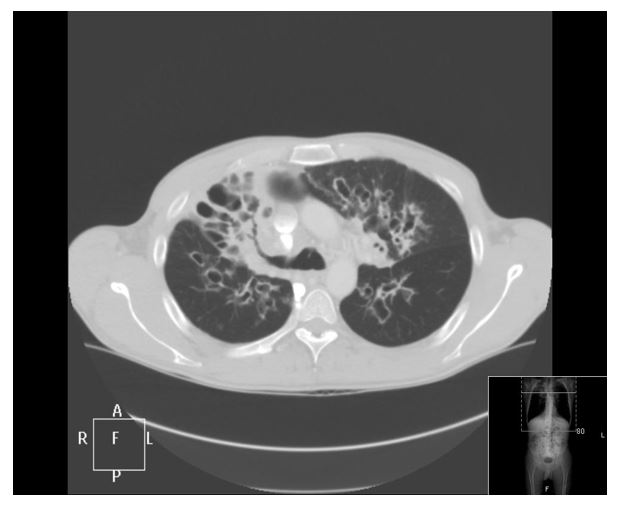
Nedre luftvägar: Det är lungorna som står för de allvarligaste symtomen. Infektioner är vanligt redan i barndomen och man vill behandla intensivt för att förhindra permanent förekomst av bakterier (kolonisation). Ökad mängd slem och påverkan på cilier medför att infektioner skadar slemhinnan, och det sega slemmet och inflammationen ger förutsättningar för kronisk påverkan med remodelling i luftvägar och påverkan på lungparenkymet med irreversibla skador som följd. Ett grundläggande problem för patienterna är att få bort det sega slemmet från luftvägarna. De flesta patienter har återkommande exacerbationer (försämringsepisoder) som kräver antibiotika (oralt, intravenöst eller som inhalation). Patienterna utvecklar över tid en kronisk obstruktivitet (dvs sjunkande FEV1 (forcerad exspiratorisk volym på en sekund)) och fysiologiskt påminner detta om förändringarna vid KOL (kroniskt obstruktiv lungsjukdom). Bronkiektasier i luftvägarna och även mer eller mindre stora lufthålor i lungparenkymet är typiskt för CF och dessa är vanligen lokaliserade i ovanloberna. Om patienter avlider före lungtransplantation beror det vanligen på respiratorisk insufficiens och komplikationer i nedre luftvägar pga infektioner.
Figur 3:
Källa Lennart Hansson
Pankreas – bukspottskörteln: I många fall är pankreasfunktionen kraftigt nedsatt redan under neonatalperioden medan den hos andra avtar under första levnadsåret och fibrotiseras (därav cystisk fibros). Man delar upp patienter i pankreassufficienta (har kvar funktionen i pankreas), som utgör 10-15 % och med vanligen mild sjukdom, och pankreasinsufficienta (saknar funktion i pankreas) som utgör 85-90 % av alla patienter (4). Nedsatt/avsaknad av pankreasfunktion medför att de inte producerar pankreasenzymer och därmed har sämre upptag av näring, fett och de fettlösliga vitaminerna A, D och E. Dessa patienter måste ta extra enzymer i samband med måltid (Creon). De med fungerande pankreas kan drabbas av pankreatiter. På ultraljud och CT buk ser man att pankreas är fibrotiskt omvandlad hos de pankreasinsufficienta patienterna.
Gallbesvär: Det är vanligt med gallbesvär tidigt hos CF-patienter. Eftersom även gallan blir segare vid CF kan det medföra leverpåverkan.
Lever: Leverpåverkan är vanligt hos patienter med CF medan allvarlig leverpåverkan är relativt ovanlig. Omkring 5-7 % utvecklar levercirros och det drabbar vanligen yngre patienter (7). Man ser ofta påverkan på levervärden men uttalad leversvikt som leder till levertransplantation är ovanligt.
Mag-tarmkanal: Det är ytterst vanligt med symtom som kan variera från lindrig gastrit/esofagit med sura uppstötningar och halsbränna till uttalade besvär från tjocktarmen. Innan man hade tillgång till pankreasenzymer var magproblem mycket besvärliga då de flesta hade uttalade diarréer pga fettmalabsorption med fettrika och täta avföringar. När Creon tillkom förbättrades situationen radikalt. Ett vanligt problem numera är förstoppningar och en speciell typ är DIOS (distalt intestinalt obstruktiv syndrom) som beror på att avföringen blir mycket seg och ger förstoppning i övergången från tunntarmen till tjocktarmen. För vissa patienter kan detta vara det dominerande och återkommande problemet. Det kan likna symtomen vid blindtarmsinflammation men det är viktigt att ha DIOS som differentialdiagnostik. Dessa patienter bör/skall inte opereras utan man försöker lösa förstoppningen med laxering. Förr var undervikt ett stort problem men introduktionen av Creon och allt bättre kostråd medför att del flesta CF-patienter upprätthåller vikten. Enstaka patienter har kvar mycket svåra buksmärtor där man inte kan påvisa orsaken trots omfattande utredning.
CF-relaterad diabetes mellitus (CFRD): Detta är en egen subtyp av diabetes vars exakta patogenes inte är klarlagd. CFRD är ett mellanting mellan typ 1 eller typ 2. Patienterna har vanligen en viss egenproduktion av insulin och utvecklar inte ketoacidos. Risken ökar med ålder och vid 30 års ålder har drygt 30 % av alla CF-patienter CF-relaterad diabetes (8). Den är speciell såtillvida att den ofta har normalt faste-blodsockervärde som sedan stiger snabbt efter födointag och snabbt går ner igen. Den skall inte behandlas med kost och kräver initialt små doser insulin. Dålig glykemisk kontroll medför ett ökat inflammatoriskt pådrag som ger större risk för infektioner och en allmänt sämre kontroll av CF-sjukdomen. Dessa patienter skall handläggas av diabetesspecialist med stor erfarenhet av CF-patienter vilket medför att det är enheter kopplade till CF-centra på Universitets-sjukhusen.
Leder: Cystisk fibros (CF) -patienter drabbas i sällsynta fall av smärtor i lederna. Det rör vanligen de stora lederna och är något avvikande då de vanligen endast presenterar sig med smärtor. De har sällan artritfynd, dvs ingen svullnad, rodnad eller värmeökning.
Osteoporos: Osteoporos är vanligt men lyckligtvis är frakturer relativt ovanligt. Med ökad rörlighet och förbättrat hälsotillstånd så kommer detta i framtiden att bli ovanligt. I Sverige ser vi i stort sett aldrig någon uttalad kyfoscolios som tyvärr var mycket vanligt tidigare.
Infertilitet: Kvinnor har en något nedsatt fertilitet pga segt cervixsekret. Många kvinnliga Cystisk fibros (CF) -patienter får barn på ”normalt” sätt medan andra behöver genomgå IVF (In Vitro Fertilisering). Vi rekommenderar att alla kvinnliga Cystisk fibros (CF) -patienter använder någon form av preventivmedel. Graviditeter hos kvinnor bör planeras väl så att de är optimalt behandlade i sin grundsjukdom. Män är i praktiken alltid infertila (inte sterila) pga avsaknad av sädesledaren (CBAVD = Congential Bilateral Abscene of Vas Deferens). Det finns några lindriga mutationer som i stort sett bara har detta som symtom och de diagnostiseras inte sällan i samband med infertilitetsutredningar. Män kan bli pappor via IVF där man tar upp spermier från testiklarna och befruktar ägget som sedan plantera in.
Malignitet: Det finns en ökad risk för malignitet, framför allt tumörer i tjocktarmen, och man rekommenderar regelbunden koloskopi från 40 års ålder.
Själ/ångest och depression: Patienter som insjuknar tidigt i ungdomen med stor påverkan av vardagen som kan innebära att de inte kan gå i skolan, till exempel inför lungtransplantation, löper stor risk att drabbas av oro och ångest. De får ofta stor inverkan på sin tonårstid med förlorade skolår och avsaknad av en normalt liv med goda vänner. Det är viktigt att uppmärksamma detta med kontakter med psykolog och kurator. Tidigt insjuknade påverkar skolgång och möjligheten för utbildning och det är viktigt med ett nära samarbete med Försäkringskassa och Arbetsförmedling.
Övrigt – Salt svett och förlust av salter men detta har föga konsekvenser i vårt klimat.
Cystisk fibros är en progressiv sjukdom dvs symtom tilltar med ålder. Med allt mer förbättrad behandling kan man upprätthålla framför allt lungfunktion, vikt och få allt mindre påverkan på lungvävnad. Med de nya läkemedlen som påverkar CFTR kan man möjligen helt förebygga kroniska förändringar.
Bakterier i luftvägarna
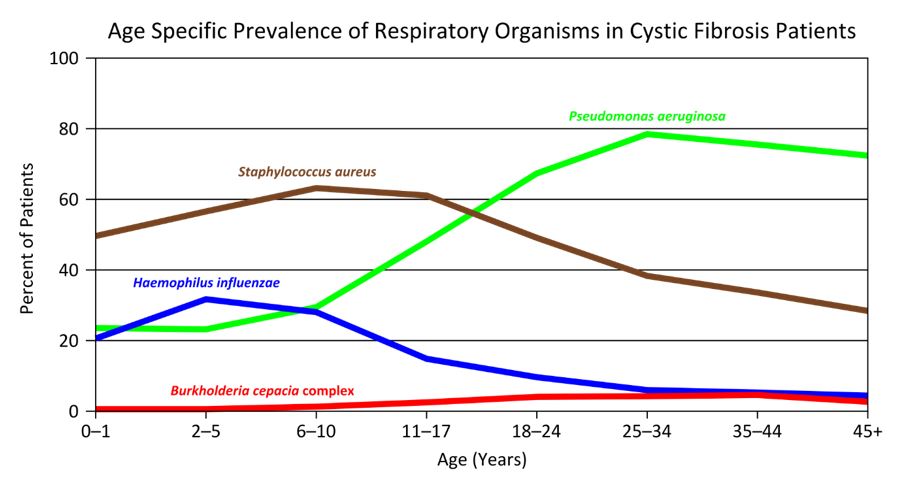
Vid födseln är lungorna sterila men så fort vi föds konfronteras vi med omvärlden och hos individer med normala luftvägar uppstår inga problem. Cystisk fibros (CF) -patienter har däremot slem och ökad klibbighet i luftvägarna och därmed fastnar bakterier. Under första åren är Stafylococcus aureus och Haemophilus influenzae vanliga. Efter hand ökar risken för infektioner med gram-negativa bakterier till exempel Pseudomonas aeruginosa som blir vanligare i tonåren (figur 4). Strävan är att försöka behandla intensivt och ofta, för att förhindra permanent växt (kolonisation) i luftvägarna. Det finns även andra bakterier t ex Burkholderia-cephacia-komplexet som betraktas som mer allvarligt. Andra bakterier är Achromobacter, Stenotrophomonas maltophilia och vissa mycobakterier och av dessa är Mycobacterium abscessus allvarligast. Svamp i luftvägarna t ex candida albicans (munsvamp) är vanligt och hos vissa patienter kan mögelsvamp, Aspergillus fumigatus vara ett problem. Med allt bättre behandlingar så koloniseras Cystisk fibros (CF) -patienter allt senare och många patienter som överförs från barncentra till vuxencentra vid 18 års ålder är inte koloniserade med Pseudomonas aeruginosa.
Diagnostik Cystisk fibros
Grunden för diagnostik är misstanke om sjukdomen baserat på symtom och kliniska fynd. Mekoniumileus vid födsel missas sällan och vid dålig tillväxt och luftvägssymtom under de första levnadsåren måste man ha CF i åtanke. Högre upp i åldern blir det klart svårare eftersom dessa patienter har färre/mindre symtom. För diagnos krävs två patologiska svettester och/eller att man påvisar två mutationer som är förenliga med CF.
Vid misstanke är svettest fortfarande grundundersökningen och den sker på ackrediterade laboratorier där man fäster små elektroder på underarmen och sänder en svag ström som stimulerar till svettbildning (pilokarpin-jontofores) där man mäter kloridinnehållet. Höga halter (>60 mmol/l) inger misstanke om CF. Svettest bör göras även om man redan har mutationsanalys. Därefter verifierar man diagnosen med mutationsanalyser, som dock inte är fullständig men täcker de vanligaste mutationerna (90 %). Om patienter kommer från andra länder kan man missa sällsynta mutationer men man kan då göra sekvensering av hela genomet för att leta efter förändringar på kromosom 7.
Om patienten har lös avföring eller dålig viktuppgång kan det vara av värde att kontrollera feces-elastas. Elastas bildas i pankreas och nedsatt värde indikerar CF. Normalt elastas utesluter inte Cystisk fibros (CF) men ett normalt värde talar emot pankreasinsufficiens.
Objektiva fynd
Röntgen: Med tilltagande sjukdom och symtom ökar risken för utveckling av bronkiektasier. Dessa ses framför allt på skivröntgen (HRCT = högupplösande CT). Man kan även följa sjukdomen med lungröntgen med tomografi.
Lungfunktion: Det är viktigt att försöka upprätthålla lungfunktionen så länge som möjligt, vilket är helt avgörande för prognosen på lång sikt. Spirometri görs vid varje kontroll på Cystisk fibros (CF) -mottagningen och en utvidgad undersökning sker i samband med årskontrollen.
LCI (lungclearance-index): LCI är ett mått att mäta lungfunktionen framför allt hos små barn. Hos vuxna har den ännu inte funnit sin plats.
Arbetsförmåga: CF-patienter följs årligen med bedömning av arbetsförmåga. Det kan ske med 6 min gångtest men om patienten mår bra ger det sällan utslag och man gör då arbetsprov på cykel med eller utan bedömning av lungfunktion (s k ergospirometri).
Odlingar från luftvägar: Sputumodlingar tas vid varje besök på CF-centrat. Man bör komplettera med svampodlingar och odlingar för tuberkulos för att kunna påvisa ev NTM (non-tuberculosis mycobactera).
Densitometri: Skelettundersökning för bedömning av benskörhet sker regelbundet men inte årligen annat än vid tilltagande osteoporos.
Glukosbelastning: Görs för att bedöma ev Cystisk fibros (CF) -relaterad diabetes mellitus.
Blodprover: CRP, vita och SR behöver inte påverkas speciellt mycket vid exacerbationer och dessa undersökningar medför ofta inte så mycket vid ställningstagande till behandling med antibiotika. Kraftigt förhöjd CRP indikerar allvarlig sjukdom. Vid septiska tillstånd med hög feber och allmänpåverkan bör man blododla.
Behandling av Cystisk fibros-patienter
Behandling av Cystisk fibros (CF) baseras på patientens egen träning, andningsgymnastik på egen hand och tillsammans med fysioterapeut samt läkemedelsbehandling (bronkvidgare, slemlösare och antibiotika). De flesta patienterna med symtom kräver inhalationer och andningsgymnastik 1-2 gånger dagligen samt egen fysisk träning flera gånger per vecka. Ju äldre patienten blir och desto sämre brukar det medföra att patienten måste lägga ännu mer tid på detta, ibland upp till flera timmar per dag i lugnt skede, med tilläggsbehandling i samband med försämringsperioder. Nästan all behandling inklusive antibiotikabehandling sker polikliniskt dvs patienten behöver inte läggas in.
Egen träning: Det är oerhört viktigt att patienterna tränar för att upprätthålla konditionen. För vissa patienter är den huvudsakliga aktiviteten som gör att slem lossar.
Andningsgymnastik: Den kan ske på egen hand eller tillsammans med fysioterapeut och innebär olika tekniker för slemmobilisering t ex huffing, PEP-träning (Positive Exspiration Pressure) och andra tekniker med syfte att lösa, mobilisera och få bort slemmet.
Läkemedel:
Luftvägar:
Vissa patienter har nytta av bronkvidgare (beta-2-stimulerare) medan andra bara får biverkningar. Man kan prova inandningskortison mot en förmodad inflammation i slemhinnan men för många har det ringa betydelse och vi överbehandlar sannolikt med inandningskortison.
Slemlösare: Tidigare användes ofta acetylcystein men det har blivit allt mindre vanligt och i stället används hypertont koksalt (3-5 %). Det finns i färdiga beredningar men patienterna blandar ofta själva från Addex®-Natriumklorid tillsammans med isotont koksalt (0,9 %) eller sterilt vatten. Hypertont koksalt kan reta luftvägarna och alla kan inte tolerera det. Även vanligt isotont koksalt som är mer skonsamt kan användas. Inandat mannitol har använts på CF-patienter och är inregistrerat men inte prisförhandlat. Pulmozyme® har hos vissa patienter mycket god effekt men är retande och kan ge besvärliga hemoptyser. Många patienter använder Bisolvon® i tablettform i ganska höga doser men det vetenskapliga underlaget är nog tveksamt. Kortison i tablettform provas ibland vid exacerbationer på samma sätt som vid KOL.
Antibiotika:
Antibiotika kan och skall ges frikostigt. Traditionerna varierar från land till land. I Danmark har man sedan 1990-talet givit mycket täta kurer med bredspektrumantibiotika. Det finns klart visat att intensifierad behandling försenar kolonisering av luftvägarna.
1. Tablettform: Många barn får flukloxacillin (Heracillin®) under vintermånader under de första levnadsåren. Haemophilus influenzae kan även behandlas med tablett t ex bioclavid. I vuxen ålder kan man prova olika varianter beroende på vilka bakterier man odlar fram. Ciprofloxacin är den enda tablettbehandlingen som kan ha effekt på Pseudomonas aeruginosa. Bactrim® i högre doser kan fungera på Stenotrophomonas maltophilia.
2. Intravenöst: Behandlingen baseras på symtom och bakterier och deras resistensmönster. Tyvärr är det vanligt att patienter utvecklar överkänslighetsreaktioner (utslag, klåda, stickningar i mun och armar) på höga doser vilket gör att man inte kan använda vissa antibiotika. Behandlingar sker i vissa fall baserat på tidsintervall, t ex var tredje månad, vilket är vanligt till barn. Andra patienter har färre försämringsperioder men det är vanligt med 2-4 kurer per år. CF-patienter tål betydligt högre doser antibiotika än icke-CF-patienter pga ökad omsättning i levern. Det kan dock leda till hörselpåverkan och balansproblem om de har fått omfattande doser av aminoglykosider.
Numera ges (åtminstone till vuxna) nästan all intravenös behandling via Homepump (figur 4) eller Intermate som tillverkas av sjukhusapotek eller privat apotek med ansvar för intravenösa beredningar. Behandlingen innebär beredning av antibiotika i små plastkuddar på 100-200 ml som kopplas till en perifer venport. Patienterna får nästan alltid två olika läkemedel vilket medför att de ska ha 4-6 pumpar per dygn. Kuren påbörjas alltid på mottagningen men sedan kan kuren, som varar 10-14 dagar, helt skötas i hemmet (eller på jobbet).
3. Inhalationsantibiotika. Det finns fyra olika typer av läkemedel (tobramycin, colistin, aztreomycin och levofloxacin) som kan inandas och de används vanligen under fyra veckor och därefter gör man uppehåll under fyra veckor före start av ny kur.
Vid nyupptäckt förekomst av Pseudomonas aeruginosa vill man försöka eradikering, dvs att behandla bort den. Då ger man först intravenös antibiotika som följs upp av tablettbehandling och inhalationsantibiotika.
Det finns ingen exakt vetenskap om hur antibiotikabehandling vid Cystisk fibros (CF) skall ske. Det gäller typ av läkemedel, intervall och vilka beredningsformer som skall användas och ofta får man kombinera olika varianter. Det kan innebära kontinuerlig tablettbehandling där man med några veckors intervall byter läkemedel eller att man har längre kurer med antibiotika-behandling eller kombinerar tabletter, inhalationer och intravenös behandling.
Bihålor: Inandat nasalt kortison kan prövas men många upplever föga lindrig. Spolning i näsan med vanligt koksalt eller instillation med antibiotika kan också prövas. Det finns en speciell nebuliseringsapparat för näsan (Pari-Sinus).
Mag-tarmkanal: Pankreasenzym (Creon) är nödvändigt för alla med pankreasinsufficiens men dosen man tar till måltider (och andra läkemedel) varierar oerhört mycket mellan individer. Många tar protonpumpshämmare (omeprazol o liknande). Fettlösliga vitaminer (A, D och E) behöver oftast kompletteras. Det är viktigt att inte bli förstoppad och tarmreglerande läkemedel måste finnas tillgängligt åtminstone vid behov. God nutrition är viktigt och kontakt med dietist är viktigt vid problem. Det är vanligt att patienter tar extra näringsdrycker men vid svåra viktproblem kan det bli aktuell med sondnäring, parenteral näring och PEG (percutan gastrostomi).
Lever: Ursofalk (ursodeoxicholsyra) används vid stigande levervärden och gör gallan mer lättflytande.
Diabetes: Insulin, ofta i mycket små doser initialt, men detta sköts bäst via erfaren endokrinolog.
Leder: Smärtstillande vid behov men vid svåra smärtor kan man behöva ge kortison i medelhög dos under kortare tid.
Benskörhet: Kalk oftast i kombination med D-vitamin. Bisfosfonat kan ges dagligen, som veckodos alt som injektion.
Extra salt: Vid resor till tropiska länder och i vistelse i hög värme med risk för vätskeförlust bör man ge extra salttabletter.
Figur 4:
Källa: Wikimedia commons
Akuta problem hos Cystisk fibros-patienter:
1. Luftvägar:
A Infektioner: Andningsbesvär som oftast är relaterade till exacerbation pga infektion/pneumoni. Patienterna kan dock utveckla en svår respiratorisk insufficiens och det är viktigt med blodgaser. Lungembolier är mindre vanligt men det måste man ändå beakta.
B Pneumothorax: Är lyckligtvis ovanligt numera men kan kräva en akut lungröntgen.
C Hemoptyser: Det är ovanligt med allvarliga hemoptyser.
2. Mag-tarmkanal:
A Pankreatit: Ses bara hos pankreassufficienta patienter. Hos de med avsaknad av pankreasfunktion ser man nästan alltid låga amylasvärden.
B DIOS: Mycket vanligt problem (se ovan).
C Ospecifika buksmärtor: Hos äldre patienter risk för kolonmalignitet.
3. Äldre Cystisk fibros (CF) -patienter:
Kan drabbas av alla vanliga akuta problem till exempel hjärtinfarkt, stroke, frakturer mm.
Narkos till Cystisk fibros-patienter:
För patienter som är välmående och med välbevarad lungfunktion är det sällan något problem med operation som kräver narkos men man får vara medveten om att det ofta kräver högre doser av till exempel smärtstillande och narkosmedel. Om patienten har uttalad nedsatt lungfunktion är det angeläget med en optimalt behandlad patient och om man ska göra elektiva ingrepp behöver man ibland förbereda med intravenös antibiotikakur och intensifierad andningsgymnastik tillsamman med fysioterapeut direkt före och efter ingreppet.
Genetisk rådgivning Cystisk fibros:
När en patient som har Cystisk fibros (CF) önskar skaffa barn är det viktigt att man partnern genomgång mutationsanalys. Detta sker med ett enkelt blodprov som analyseras vid ett kliniskt genetiskt laboratorium. Dessa kan även ställa upp med ytterligare genetisk rådgivning. Om det skulle visa sig att partnern är bärare (då blir risken 50 % att få ett barn med CF), kan man genomgå IVF och testa det befruktade ägget för CF-mutationer innan man inplanterar det, så kallad Preinplantatorisk genetisk diagnostik (PGD).
Nya behandlingar – CFTR-modulatorer:
För cirka 10 år sedan kom de första läkemedlen som kunde påverka CFTR och idag finns fyra läkemedel som är godkända i USA och Europa, men endast ett av dem är tillgängligt i Sverige. Först introducerades ivakaftor som kan förskrivas till patienter som bär mutationen G551D och sju andra sällsynta mutationer, från 4 månaders ålder. Patienterna blev klart bättre men i Sverige har vi få patienter med dessa mutationer. När sedan kombinationsläkemedlet ivacaftor/lumacaftor introducerades kunde man behandla patienter som är homozygota för delF508 och äldre än 2 år vilket i Sverige utgör c:a 50 % av alla patienter. Många blev bättre men hade måttlig effekt på lungfunktion och biverkningar i form att tryckkänsla över bröstet och magbesvär var vanligt. Tezakaftor/ivakaftor har liknande effekt och indikation som ivacaftor/lumacaftor men mindre biverkningar men är tillgängligt endast på licens i Sverige.
Det senaste läkemedlet som godkänts är elexakaftor/tezakaftor/ivakaftor som inkluderar även patienter med enbart en mutation av typ delF508, vilket skulle öka andelen behandlingsbara individer upp till 85 % av CF populationen (2). I USA och i stora delar av Europa finns nu trippelkombinationen elexakaftor/tezakaftor/ivakaftor att tillgå och den har medfört stora förbättringar på lungfunktion, svettest, vikt och symtom och därmed stora förhoppningar om att kunna behandla mycket tidigt. Det används i flera länder och förhandlingar pågår om att kunna skrivas ut i Sverige, där för närvarande endast ivacaftor/lumacaftor är tillgängligt. Det utvecklas hela tiden nya kaftorer som sannolikt kommer att ytterligare förbättra vården i framtiden.
Prognos Cystisk fibros
Livslängden har kraftigt ökat under de senaste 20-30 åren. På 1960-talet nådde ytterst få patienter vuxen ålder och medellivslängden på 1980-talet var 26 år vilket numera är medianåldern på hela CF-populationen i Sverige. Den förväntade medelåldern är nu minst 50 år och kommer sannolikt att öka kraftigt inte minst vid tillgång till nya behandlingar (9).
Lungtransplantation
Lungtransplantation har varit den sista utvägen vid uttalade symtom från lungorna. När FEV1 brukar närma sig 30 % av förväntat värde brukar man ange att det är dags att påbörja utredningen inför lungtransplantation, men det finns många faktorer som spelar in. I Sverige har vi lungtransplanterat c:a 125 patienter sedan 1992. Vi har mycket goda överlevnadssiffror internationellt sett med en medianöverlevnad på c:a 14 år (10). I och med introduktionen av kaftorerna ser man i USA ett klart minskat behov av lungtransplantation och data från Europa visar att många patienter som står på väntelistan kan tas bort.
Nyföddhetsscreening för cystisk fibros
I många länder gör man nyföddhetsscreening för cystisk fibros. Det sker på samma blodprov som man undersöker för PKU (PhenylKetonUri), som är en mycket allvarlig sjukdom som leder till utvecklingsstörning om det inte diagnostiseras, men som kan förhindras med kostråd om man undviker kost med fenylketoner. För cystisk fibros så undersöker man förekomst av immunoreaktivt tryptogen (IRT) som är förhöjda hos nyfödda barn med Cystisk fibros (CF). Det finns dock en risk för överdiagnostik (falskt positiv svar) varför man måste verifiera diagnosen med mutationsanalys och svettest. Eftersom man med mutations-analyser får med alla kända varianter, kan det innebära att man tidigt (1-2 mån ålder) diagnostiserar en sjukdom som kommer att ge väldigt ringa symtom och ev endast problem med att få barn (fertilitet). Det har inneburit att man i vissa länder inte har meddelat dessa mycket lindriga mutationer till föräldrar för att man inte i onödan ska oroa dem. Sverige är ett av mycket få länder i Europa där man inte har nyföddhetsscreening. Svenska Cystisk fibros (CF) -läkare har i många år önskat att införa nyföddhetsscreening men Socialstyrelsen har vid flera tillfällen avslagit detta med hänvisning till att det är etiskt problematiskt.
Handläggning av Cystisk fibros-patienter:
CF-patienter i Sverige handläggs vid ett av fyra Cystisk fibros (CF) -centra (Lund, Göteborg, Stockholm (Huddinge) och Uppsala. Fram till 18 års ålder sköts de via barncentra och därefter flyttas de över till vuxencentra (Huddinge har ett gemensamt centrum för barn och vuxna). Patienter besöker centra allt från varje månad till en gång om året beroende på hur svårt sjuk patienten är. I många fall (framför allt barn) sköts de i samråd med barnkliniker på hemorten (shared care). Det är viktigt att patienterna handläggs gemensamt i team där de träffar läkare, sjuksköterska, fysioterapeut, dietist, psykolog och kurator. Ofta kopplar man in andra specialister som gastroenterologer, diabetes- och öronläkare.
Framtid Cystisk fibros
Från att ha varit en barnsjukdom där få patienter för 50 år sedan nådde vuxen ålder är Cystisk fibros (CF) idag en vuxensjukdom med allt bättre behandlade patienter tack vare högspecialiserade CF-centra, bättre kost, bättre andningsgymnastik och allmän träning, bättre antibiotika och förbättrade inhalationsläkemedel. Med introduktionen av de nya CFTR-modulatorerna och nyföddhetsscreening kan vi möjligen se fram emot en tid när patienterna får behandling redan innan de utvecklar symtom och därmed kanske aldrig får några svåra luftvägssymtom och kan leva nästan ett normalt liv.
Lennart Hansson
Överläkare, Med Dr
Cystisk fibrosmottagningen
Skånes Universitetssjukhus
Referens
- cff.org/managing-cf/understanding-changes-life expectancy 2022.
- ECFS Patient Registry – Annual Data Report. ECFSPR Annual Report 2019, Orienti A, Zolin, A, Jung A, van Rens J et al 2021.
- cff.org/intro-cf/about-cystic fibrosis
- Socialstyrelsen – sällsynta diagnoser – cystisk fibros. 2022. www.socialstyrelsen.se/sallsynta-halsotillstand
- Burgel et al. Future trends in cystic fibrosis demography in 34 European countries. Eur Respir J. 2015;46:133-141.
- Farrel, PM. The prevalence of cystic fibrosis in the European Union. Journal of Cystic Fibrosis. 2008;7(5):450-3.
- Leung, DH, Narkewicz. Cystic Fibrosis-related cirrhosis. Journal of Cystic Fibrosis. 16: Suppl 2: 2017: S50-61.
- Olesen HV, Drevinek P, Gulmans VA et al. Cystic fibrosis related diabetes in Europe: Prevalence, risk factors and outcome. Journal of Cystic Fibrosis. 2020;19:321-327.
- Balfour-Lynn IM, King JA. CFTR modulator therapies – Effect on life expectancy in people with cystis fibrosis. Paediatri Respir Rev 2020: May 26;S1526-0542 (20)30081-6.
- Gilljam M, Nyström U, Dellgren G, Skog I, Hansson L. Survival after lung transplantation for cystic fibrosis in Sweden. Eur J Cardiothorac Surgery 2017 March 1:51(3):571-576.