Aktuellt

Sammanfattning av lungan genom livet – en nationell utbildningsdag
Vilka faktorer från födelse, eller kanske ännu tidigare, till vuxen ålder spelar roll för de tillstånd och sjukdomar som senare kan leda till KOL?
Läs mer
Inhalationsteknik är en färskvara
En studie med svenska KOL-patienter från 2021 visade att totalt 66 procent av patienterna i studien inte tog sin inhalation på rätt sätt och att ju fler inhalatorer patienterna hade,
Läs mer
Patientfall med kangrelor
Här kan du ta del av tre intressanta patientfall med kangrelor, framtagna i samarbete med överläkaren och interventionskardiologen Per Grimfjärd, MD, PhD vid Region Västmanland (engelskt tal). Gå till Kardiologi
Läs mer
KOL och träning
Rädsland och obehaget att bli andfådd kan göra att patienter med KOL avstår från att anstränga sig. Din kunskap och råd från dig om att fysisk träning minskar risken för
Läs merWebinarer

Exacerbationer
Webbinarium KOL – exacerbationer Varför är det viktigt att undvika exacerbationer? Vilka verktyg kan man använda sig av för att bedöma grad av exacerbation? Är dyspné en prediktor för kommande
OBS utbildningsserie
Se mer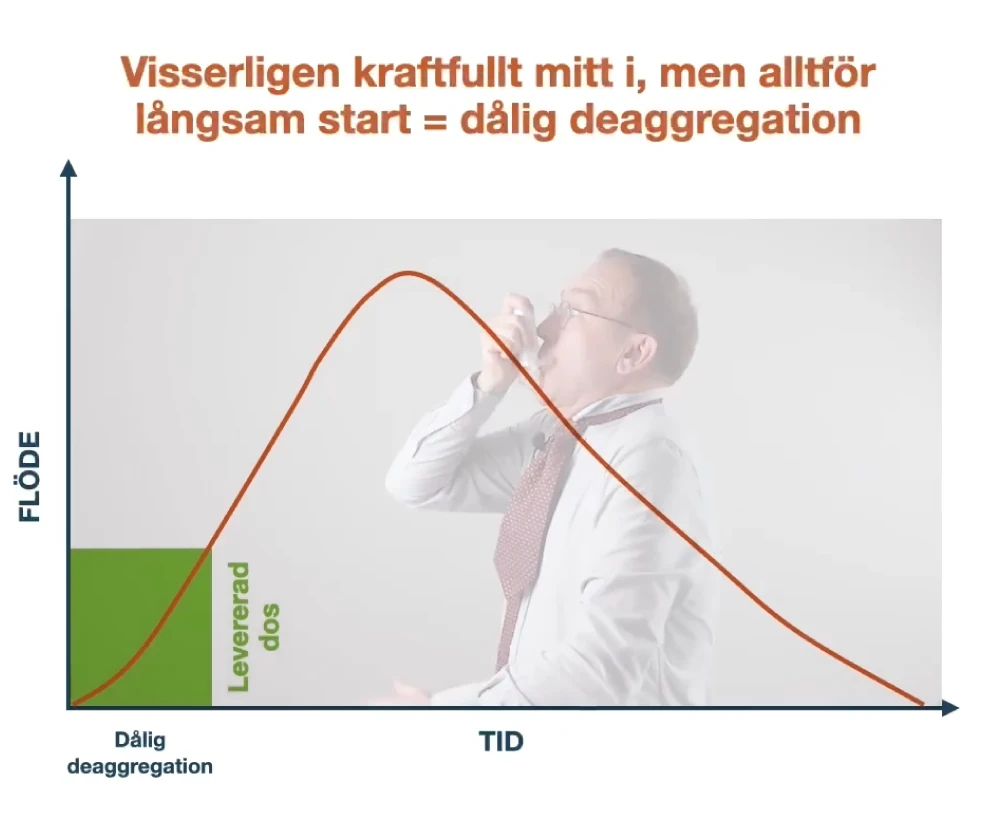
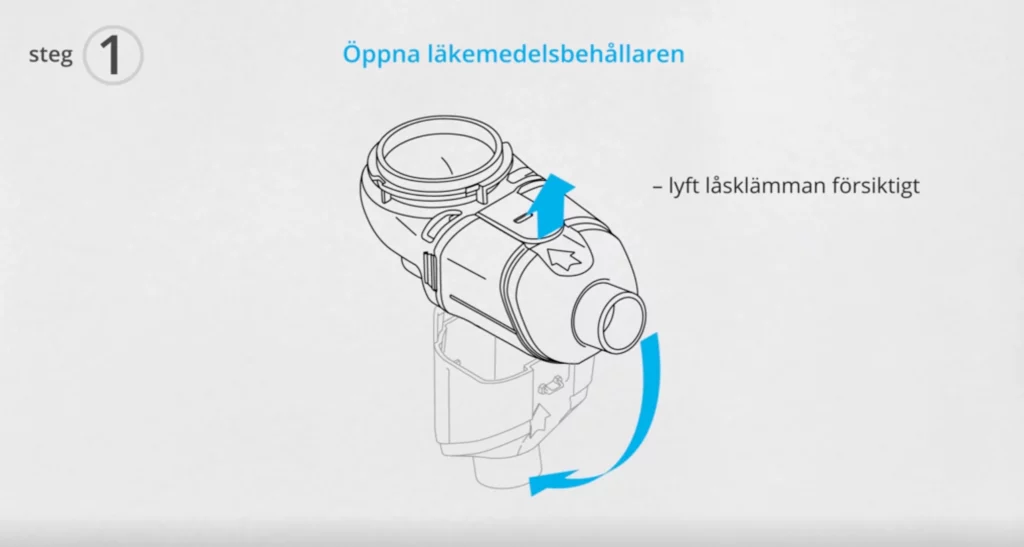
Inhalationsteknik
Att kunna inhalera rätt är en viktig faktor för att få bästa möjliga effekt av inhalerade astma- och KOL-läkemedel.
Läs mer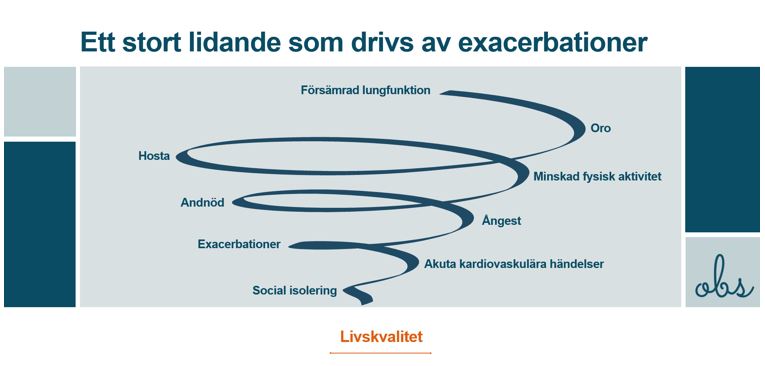
KOL-behandlingens ABE
Nu uppdaterad! Visste du att omkring en halv miljon svenskar lever med KOL?
Läs mer
KOL – sanning eller myt
Är KOL detsamma som en dödsdom? Får man automatiskt KOL om man har astma?
Läs merInstruktionsfilmer
Se alla Instruktionsfilmer